Історія Бенджаміна Баттона, народженого старим і молодіючого з роками, зачаровує уявою мільйонів завдяки новелі Ф. Скотта Фіцджеральда та однойменному фільму з Бредом Піттом. Цей мотив народжує безліч питань про можливість зворотного старіння. У реальності жодне захворювання не відтворює точне зворотне старіння, проте народна назва «хвороба Бенджаміна Баттона» міцно закріпилася за синдромами прискореного старіння, насамперед за прогерією Хатчинсона-Гілфорда (HGPS). Діти з цією патологією виглядають і відчувають себе набагато старшими за свій хронологічний вік, стикаючись із симптомами, типовими для літніх людей.
Прогерія — надзвичайно рідкісне генетичне захворювання, що змушує організм старіти в прискореному темпі. Більшість дітей народжуються з нормальним виглядом, але вже у перші роки життя з’являються зморшки, випадіння волосся, проблеми з суглобами та серцево-судинною системою. Середня тривалість життя без сучасного лікування — близько 14–15 років, переважно через ускладнення атеросклерозу. Сучасні терапії, такі як лонафарніб, змогли подовжити це до майже 20 років у деяких випадках.
Іноді термін плутають із хворобою Баттена (нейрональними цероїдними ліпофусцинозами), яка також вражає дітей і призводить до тяжких неврологічних проблем, але механізми та прояви відрізняються. Розуміння цих станів допомагає не лише розвіяти міфи, а й підтримати реальних пацієнтів та їхні родини.
Літературне коріння та культурний вплив
Новела Фіцджеральда 1922 року описує життя чоловіка, який починає шлях у тілі немовляти з рисами старця і поступово «молодіє». Фільм 2008 року розвинув цю ідею, додавши емоційну глибину стосункам і суспільним викликам. Ці твори торкаються тем часу, ідентичності та самотності, роблячи «зворотне старіння» потужною метафорою людського існування. Багато глядачів після перегляду шукають інформацію про реальні аналоги, що призвело до поширення народної назви для прогерії.
Культурний ефект виявився двояким: з одного боку, він підвищив обізнаність про рідкісні захворювання, з іншого — створив плутанину. Реальні пацієнти з прогерією часто стикаються з упередженнями чи надмірною увагою саме через асоціацію з фільмом. Водночас історії таких людей, як Сем Бернс, який жив з прогерією до 17 років і надихав інших своєю позитивною філософією, демонструють силу духу.
Що таке прогерія насправді: наукове пояснення
Класична прогерія Хатчинсона-Гілфорда виникає через мутацію в гені LMNA, який кодує білок ламін А. Цей білок формує каркас ядра клітини. Мутація призводить до накопичення аномального білка — прогерину, який дестабілізує ядерну оболонку, викликає хронічне запалення, пошкодження ДНК та прискорену загибель клітин. Організм починає «зношуватися» в 5–10 разів швидше.
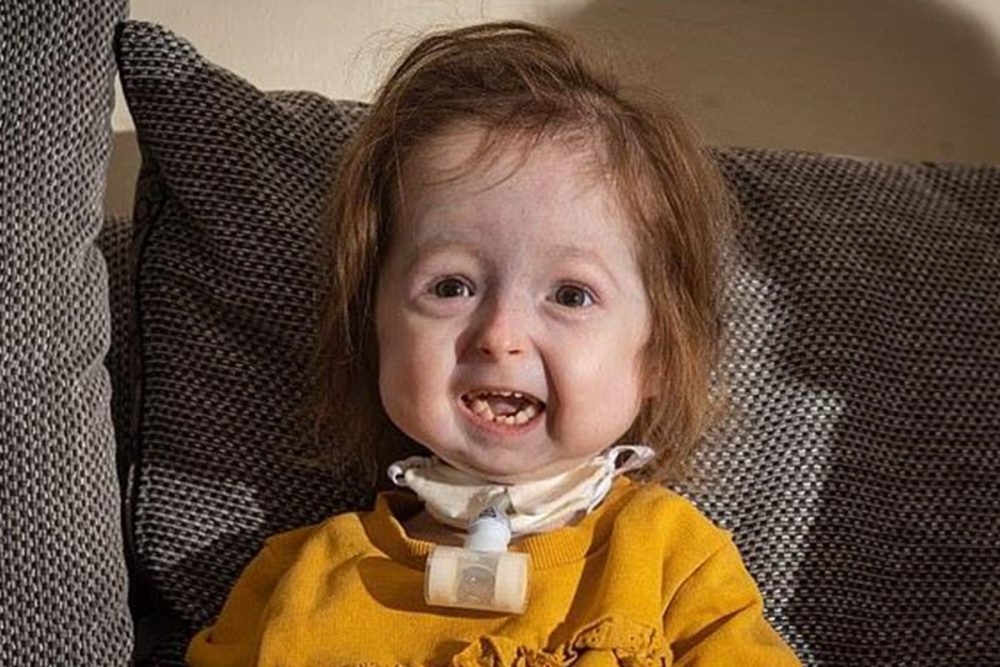
На відміну від нормального старіння, процес тут запускається рано і не залежить від способу життя. Діти виглядають здоровим при народженні, але до 1–2 років з’являються характерні ознаки: уповільнення росту, втрата підшкірного жиру, зморшкувата шкіра, великі очі, маленький підборіддя, «дзьобоподібний» ніс. Пізніше розвивається алопеція, проблеми з зубами, тугоподвижність суглобів, остеопороз і тяжкий атеросклероз.
Інші форми проgerоїдних синдромів, наприклад синдром Вернера, проявляються пізніше — у дорослих, і пов’язані з іншими генетичними дефектами, що впливають на ремонт ДНК. Вони теж викликають передчасне старіння, але з іншими нюансами, як ранній розвиток діабету чи катаракти.
Симптоми та перебіг захворювання: етапи життя дитини з прогерією
Перші прояви часто непомітні: повільне набір ваги та росту. До двох-трьох років формується типовий зовнішній вигляд — тонка шкіра з венами, відсутність волосся на тілі та голові, обмежена рухливість. Діти залишаються розумово розвиненими, але фізичні бар’єри серйозно впливають на життя.
Серцево-судинні проблеми стають головною загрозою: артерії тверднуть, як у людей похилого віку, підвищуючи ризик інфаркту чи інсульту. Суглоби втрачають гнучкість, кістки стають крихкими. Слух може погіршуватися, зір — страждати від вторинних ускладнень. Незважаючи на це, багато дітей ведуть активне соціальне життя, відвідують школу, займаються творчістю.
Перебіг індивідуальний, але без втручання більшість пацієнтів не доживають до 20 років. Сучасні підходи змінюють цю статистику.
Діагностика та лікування: від генетичного тесту до терапії
Діагноз підтверджується генетичним тестом на мутацію LMNA. Раннє виявлення критично важливе для початку підтримки. Лікування симптоматичне: фізіотерапія для суглобів, кардіологічний моніторинг, спеціальне харчування. У 2020-х роках FDA схвалила лонафарніб (Zokinvy) — інгібітор, який блокує фарнезилювання прогерину, зменшуючи його токсичність. Це подовжує життя на кілька років і покращує якість.
Дослідження генної терапії, стовбурових клітин та інших молекул тривають. Батьки та фонди, як Progeria Research Foundation, активно підтримують науку.
Порівняння з хворобою Баттена та іншими станами
Хвороба Баттена (нейрональні цероїдні ліпофусцинози) — група лізосомних хвороб накопичення, коли в клітинах, особливо нейронах, накопичуються токсичні речовини. Вона призводить до втрати зору, судом, деменції та ранньої смерті, але не викликає класичного «старіння зовнішності», як прогерія. Плутанина в назвах походить від схожості рідкісності та дитячого початку.
Інші рідкісні стани, як мандибулоакральна дисплазія, теж можуть давати ефект «зупинки росту» чи передчасних змін, але механізми інші.
| Аспект | Прогерія (HGPS) | Хвороба Баттена |
|---|---|---|
| Основна причина | Мутація LMNA, прогерин | Мутації генів CLN, накопичення ліпофусцину |
| Головні симптоми | Прискорене старіння зовнішності, серцево-судинні проблеми | Втрата зору, судоми, неврологічний регрес |
| Тривалість життя | Переважно до 15–20 років | Залежно від форми, часто до 10–20 років |
| Лікування | Лонафарніб та симптоматичне | Симптоматичне, деякі форми — ферментна замісна терапія |
Дані таблиці базуються на оглядах StatPearls та клінічних описах (NCBI, MedAboutMe).
Цікаві факти
Прогерія трапляється приблизно в 1 випадку на 4–20 мільйонів народжень. У світі живе кілька сотень діагностованих дітей. Деякі пацієнти досягають 20+ років завдяки терапії. Дослідження прогерії допомагає зрозуміти нормальне старіння, адже механізми частково перетинаються з віковими змінами в клітинах.
Історії конкретних людей надихають: від Сем Берна, який грав на барабанах і мріяв про космос, до сучасних підлітків, що ведуть блоги та борються за обізнаність. Ці діти часто демонструють надзвичайну зрілість і оптимізм.
Життя з діагнозом: емоційні, соціальні та практичні аспекти
Родинам доводиться стикатися з важким емоційним навантаженням — від шоку діагнозу до щоденної турботи. Діти потребують адаптованого середовища, підтримки в освіті та соціалізації. Багато хто відвідує звичайні школи з асистентами, займається мистецтвом чи спортом, адаптованим під можливості.
Суспільство грає ключову роль: толерантність, освіта про рідкісні хвороби зменшують стигму. Фонди надають ресурси, організовують зустрічі сімей. У 2025–2026 роках прогрес у генній терапії та персоналізованій медицині дає надію на кращі результати.
Для батьків важливо шукати спеціалістів, приєднуватися до спільнот і фокусуватися на якості життя тут і зараз — маленьких радощах, досягненнях дитини, міцних сімейних зв’язках.
Майбутнє: надії науки та підтримки
Сучасні дослідження спрямовані на корекцію мутації, очищення від прогерину та захист клітин. Клінічні випробування дають обнадійливі результати. Паралельно розвивається паліативна допомога, яка покращує комфорт.
Підвищення обізнаності через медіа, документальні фільми та соціальні мережі прискорює збір коштів і залучення вчених. Кожна історія пацієнта додає мотивації для прогресу.
Хвороба, яку називають «хворобою Бенджаміна Баттона», нагадує про крихкість часу та цінність кожного моменту. Вона спонукає цінувати здоров’я, підтримувати рідкісних воїнів і продовжувати науковий пошук, який одного дня може змінити правила гри.